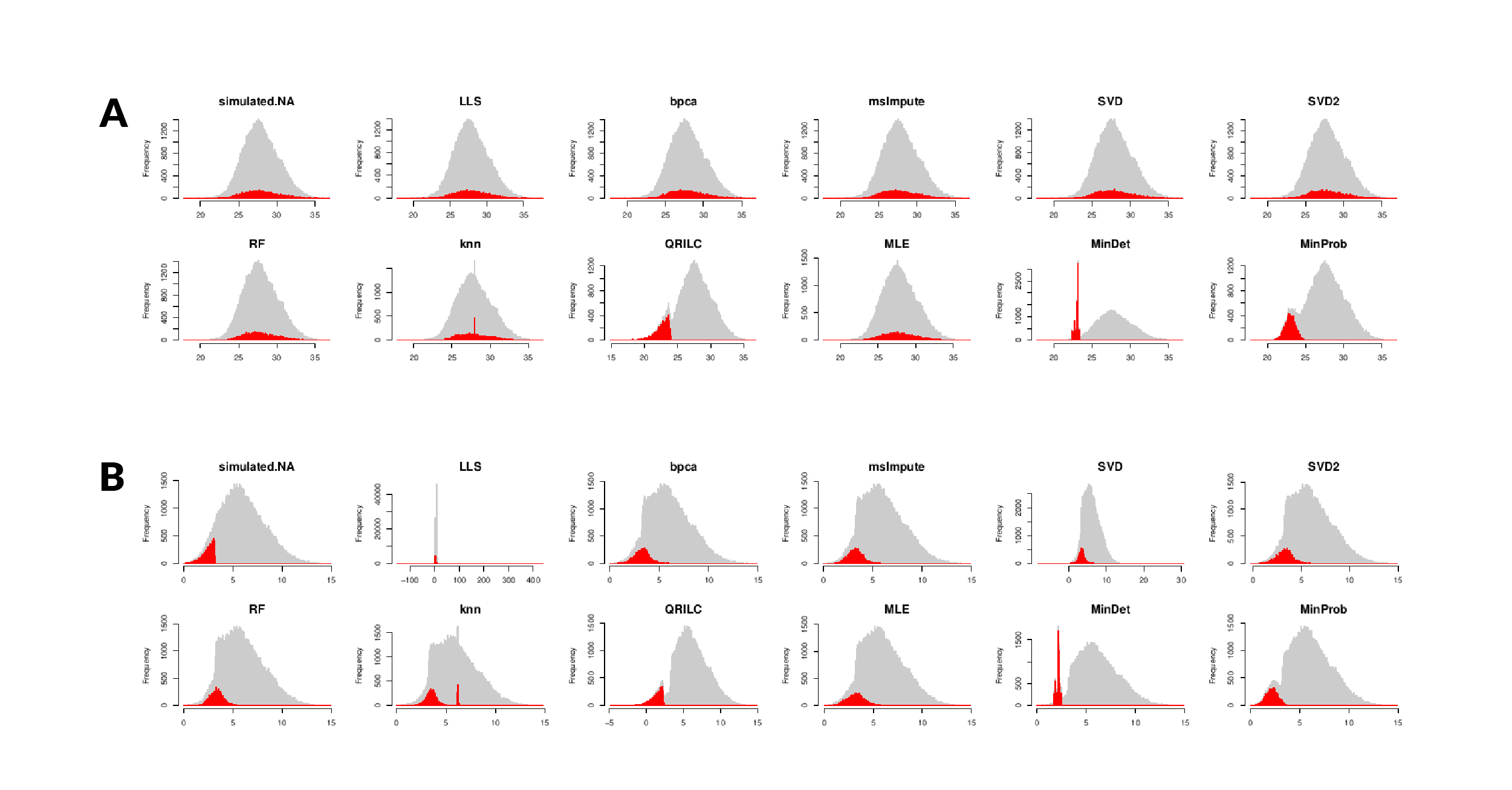
MVs in data matrices make problematic principal component analysis, batch correction and clustering. Therefore, MVs reduce discovery power and should be accurately ‘imputed’.
Previous works have compared different imputation methods.
For example, Jin et al. (1), compared LOD, ND, kNN, LLS, RF, SVD and BPCA. They concluded that RF and LLS are the best performing imputing methods, and that BPCA outperforms SVD. The MNAR ratio affected the accuracy of all datasets.
Wei et al. (2), compared performance and accuracy of 8 distinct methods in metabolomics data, including half-minimum, zero, mean, median, RF, SVD, kNN and QRILC. They also recommended RF as the best imputing method for MAR, while QRILC to be preferred for left-censored MNAR MVs.
Wang et al. (3), integrated 23 imputation methods into the R package NAguideR and evaluated their performance in proteomic data. Differently to previous studies, they found that BPCA and KNN-based rank among the top methods. In summary, RF, LLS, BPCA, and KNN are often reported as top-ranking methods. However, they are also often among the slowest.
Ideally, the imputation method of missing values in proteomics should be tailored to the nature of MVs. For instance, left-censored MNAR could be imputed with left-censored-specific methods like LOD and ND, while MAR imputed with RF and LLS. However, this is of difficult application for real world datasets where is difficult to disentangle MAR and MNAR.