
Partial clustering according to whether mice had been exposed to social interactions (social) or not (naive) could be observed using UMAP. However, no clear separation by microbiome and treatment state could be observed (Fig. 1).

Next, I performed a pairwise gene expression analysis, as shown in Table 1. Unlike the original article, I used the intersection of three differential expression analysis algorithms (Deseq2, EdgeR and limma) to identify significantly dysregulated genes (FDR<0.1, logFC>|±0.5|), which yielded a lower number of hits. The GF-SI vs GF comparison had the highest number of dysregulated genes, followed by the CON-SI vs CON comparison (Fig. 2), consistent with the findings obtained by the authors.
| Samples Group | Reference Samples Group |
| GF | CON |
| exGF | CON |
| GF | exGF |
| CON-SI | CON |
| GF-SI | GF |
| exGF-SI | exGF |
| GF-SI | CON-SI |
| exGF-SI | CON-SI |
| GF-SI | exGF-SI |
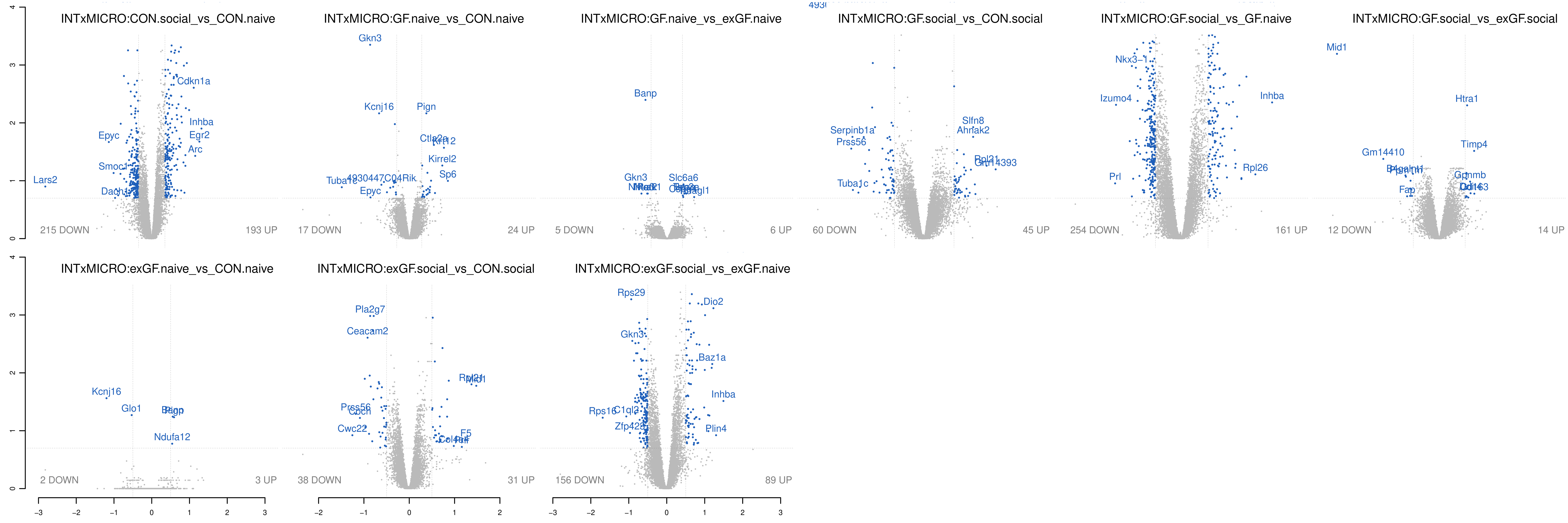
An intersection of of the comparisons involving GF and CON groups (GF-SI vs GF, CON-SI vs CON, GF vs CON and GF-SI vs CON-SI) revealed a number of genes that were activated or repressed upon exposure to social stimuli (51 UP and 17 DOWN), regardless of microbiome state. However, there were significant differences in dysregulated genes between control (101 UP and 103 DOWN) and germ-free comparisons (94 UP and 207 DOWN) (Fig. 3).

Analysis of GO terms and of KEGG pathways confirmed the main findings of the authors (Fig. 4). There was an upregulation of pathways associated with the splicesome machinery, regulation of gene expression and protein folding in GF mice after exposure to social interactions. I also observed an upregulation of the MAPK pathway in control mice following social stimulation (Fig. 5).


Furthermore, by using the geneset enrichment module in the platform, I could rapidly generate GSEA plots for the “Splicesome” KEGG pathway and the ”RNA splicing” GO term, replicating the results from the original manuscript (Fig. 6A-B).

The gene perturbation L1000 database available on the platform can be used to explore how the expression changes in a single gene correlate with an experimental gene expression profile. I used it to identify if the expression or downregulation of an individual gene could be used to understand more about the MOA of microbiome removal on social interactions using the GF-SI vs CON-SI pairwise comparison.
Repression of transcription factor ETS2 (ERF) was found to mimic the differences in gene expression observed between GF-SI and CON-SI (Fig. 7A). ERF may be needed for correct neuronal function (Maroulakou and Bowe, 2001) and downregulation has been observed in subjects with autism spectrum disorder (Burraco et al, 2016). This appears consistent with the information provided by the platform.
A

B

Figure 7. GSEA plots obtained by comparing the GF-SI vs CON-SI signature against the L1000 individual gene perturbation database. (A) ERF gene silencing, (B) EDN3 expression inhibition.
Conversely, repression of EDN3 (endothelin 3) expression was found to generate an opposite profile to the one observed for the GF-SI vs CON-SI pairwise comparison (Fig. 7B). Endothelins act as neurotransmitters in the central nervous system and EDN3 has been observed to be upregulated in people suffering from autism spectrum disorder (Burraco et al, 2016). This result would thus suggest that upregulation of EDN3 may play a role in impaired social behaviour in GF-SI mice.
Finally, I explored drugs that could inhibit the gene expression profile generated by stimulated GF mice against stimulated CON mice. The main mode of action (MOA) among inhibitory drugs was mTOR pathway inhibition (Fig. 8).
Interestingly, suppression of the AKT/mTOR pathway has been shown to rescue social behaviour in genetically modified mice (Xing et al, 2019), while increased mTOR pathway activation has been associated with autism (Rosina et al, 2019). The list also includes PI3K inhibitors, with the PI3K pathway strongly intertwined with the AKT/mTOR pathway.
The second most common family of inhibitors, glycogen synthase kinase (GSK) inhibitors, is also interesting, since GSK3 overexpression has been shown to influence social preference and induce anxiety during social interactions (Mines et al, 2010) and GSK3 inhibition is recognised as a treatment for psychiatric disorders (Finkelman and Martinez, 2011).



Axel Martinelli’s academic background is in molecular biology and parasitology. He earned a Ph.D. on the genetics of strain-specific immunity against malaria infections and a master’s degree in bioinformatics with specialization in the analysis of omics data. During his postdoctoral career, he worked on genomics and transcriptomics studies and is currently the head of biology at Bigomics Analytics.